Private Label, White Label, Wholesale partnerships available - EU, USA and UK - Free shipping from €75
Définition de la pureté pharmaceutique : guide chercheurs
Découvrez la définition de la pureté pharmaceutique : un guide essentiel pour les chercheurs, garantissant contrôle de qualité et conformité.
TL;DR:
- La pureté pharmaceutique est le rapport entre la concentration mesurée d’une substance active et sa valeur théorique, selon un profil multi-paramétrique. Elle dépend du contrôle rigoureux des impuretés, identifiées et quantifiées par des méthodes analytiques validées comme la HPLC. La conformité des lots repose sur les seuils réglementaires fixés par la Ph. Eur. et les lignes ICH, notamment pour les impuretés organiques, inorganiques, solvants, mutagènes et N-nitrosamines.
La pureté pharmaceutique se définit comme le rapport entre la concentration mesurée d’une substance active et sa concentration théorique attendue, exprimé en pourcentage selon la formule %pureté = (Cexp/Ctheo) × 100. Cette définition opérationnelle, encadrée par la Pharmacopée Européenne (Ph. Eur.) et les lignes directrices ICH, est indissociable du contrôle rigoureux des impuretés et des méthodes analytiques validées comme la HPLC. Pour les chercheurs et universitaires en sciences pharmaceutiques, comprendre cette définition précise constitue le socle de toute démarche de contrôle de qualité pharmaceutique fiable et reproductible.
Qu’est-ce que la définition de la pureté pharmaceutique ?
La pureté pharmaceutique ne se réduit pas à un chiffre global. Elle traduit la conformité d’une substance active par rapport à un profil d’impuretés défini et contrôlé, selon des spécifications établies par type d’impureté plutôt que par une valeur unique agrégée. Cette nuance est fondamentale : deux lots peuvent afficher une teneur identique en principe actif mais présenter des conformités différentes selon la nature des impuretés critiques détectées.

L’EDQM (European Directorate for the Quality of Medicines) relie explicitement la pureté au contrôle des impuretés, en la cadrant par les monographies de la Ph. Eur. et les lignes directrices ICH Q3A, Q3B et Q3C. Cette approche multi-paramétrique signifie qu’un résultat analytique isolé ne suffit jamais à qualifier un lot. La pureté et l’efficacité des médicaments sont donc liées à la capacité des laboratoires à détecter, identifier et quantifier chaque classe d’impuretés avec des méthodes validées.
En pratique, les analyses des principes actifs doivent être fiables, spécifiques et validées pour démontrer la pureté et la conformité réglementaire d’un lot. Une impureté de synthèse non détectée peut fausser la quantification et conduire à la libération d’un lot non conforme, avec des conséquences directes sur la sécurité des patients.
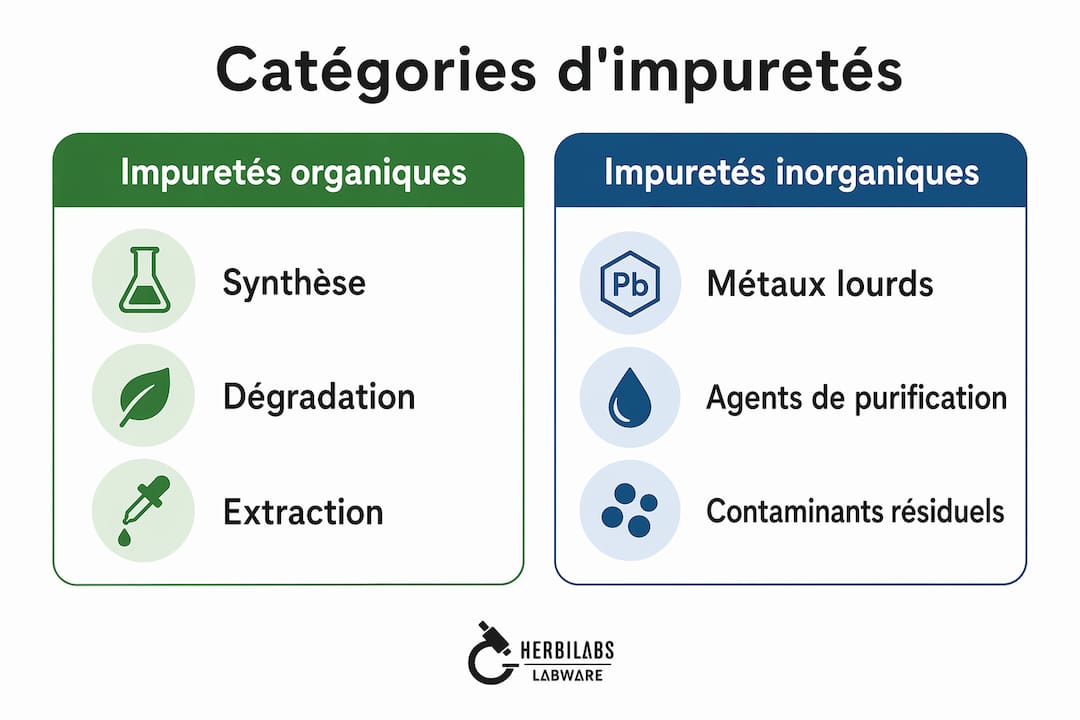
Quelles sont les catégories d’impuretés en pharmaceutique ?
Le contrôle des impuretés constitue le cœur opérationnel de la définition de la pureté pharmaceutique. Les normes de pureté pharmaceutique distinguent plusieurs grandes catégories, chacune soumise à des exigences analytiques et réglementaires spécifiques.
Les principales catégories d’impuretés reconnues par la Ph. Eur. et les lignes ICH sont :
- Impuretés organiques : produits de dégradation, intermédiaires de synthèse, sous-produits réactionnels. Elles sont souvent les plus toxicologiquement préoccupantes et nécessitent une identification structurale au-delà de certains seuils de qualification.
- Impuretés inorganiques : métaux lourds, catalyseurs résiduels, réactifs minéraux. Leur contrôle repose sur des techniques comme l’ICP-MS (spectrométrie de masse à plasma à couplage inductif).
- Solvants résiduels : encadrés par l’ICH Q3C, classés en trois classes selon leur toxicité. Le dichlorométhane ou le méthanol, par exemple, font l’objet de limites strictes exprimées en ppm.
- Impuretés DNA-réactives (mutagènes) : encadrées par l’ICH M7, elles exigent une évaluation du risque génotoxique et des limites d’exposition journalière calculées selon le concept TTC (Threshold of Toxicological Concern).
- N-nitrosamines : catégorie émergente depuis les alertes sur le valsartan en 2018, intégrée dans le contrôle des impuretés Ph. Eur. avec des procédures analytiques dédiées et des seuils d’acceptabilité très bas (souvent de l’ordre de quelques dizaines de ng/jour).
Chaque catégorie influence différemment la sécurité et la toxicité du médicament. Une impureté organique à 0,1 % peut être acceptable pour une molécule à faible potentiel toxique, mais totalement inacceptable pour un composé génotoxique potentiel. Cette gradation du risque est précisément ce qui rend la définition de la pureté pharmaceutique plus complexe qu’un simple pourcentage.
Comment mesure-t-on la pureté pharmaceutique ?
La mesure de la pureté repose sur des méthodes analytiques quantitatives validées, dont la HPLC (chromatographie liquide haute performance) représente la référence dominante pour les principes actifs. Le principe de l’étalonnage externe, fondé sur la loi de Beer-Lambert, permet de relier l’aire de pic chromatographique à la concentration de la substance analysée.
Le calcul du pourcentage de pureté suit la formule établie :
- Préparation de l’étalon de référence : une solution de concentration théorique connue (Ctheo) est préparée à partir d’un étalon certifié, idéalement un étalon de référence primaire de la Ph. Eur. ou de l’USP.
- Analyse de l’échantillon : la concentration expérimentale (Cexp) est déterminée par comparaison de l’aire de pic de l’échantillon avec celle de l’étalon, en appliquant un facteur de réponse calculé.
- Calcul du pourcentage de pureté : %pureté = (Cexp/Ctheo) × 100. Un résultat de 99,5 % signifie que 0,5 % de la masse analysée correspond à des impuretés ou à de l’eau résiduelle.
- Validation de la méthode : spécificité, linéarité, exactitude, précision et robustesse doivent être démontrées selon les critères ICH Q2(R1) avant toute utilisation en contrôle de lot.
- Interprétation des résultats : le résultat brut est comparé aux spécifications de la monographie ou du dossier d’enregistrement pour statuer sur la conformité.
Une mauvaise quantification, due par exemple à une dérive de l’étalon ou à une interférence chromatographique non détectée, peut conduire à une surestimation de la pureté. Ce risque est particulièrement critique pour les substances à faible marge thérapeutique. La fiabilité des méthodes analytiques conditionne directement la validité des décisions qualité prises sur les lots.
Conseil de pro: Lors de la validation d’une méthode HPLC pour le contrôle de pureté, vérifiez systématiquement la stabilité de votre étalon de référence en solution sur la durée d’analyse complète. Une dégradation de 0,3 % de l’étalon pendant une séquence de 8 heures peut invalider l’ensemble des résultats de pureté du lot.
Quelles normes réglementaires gouvernent la pureté pharmaceutique ?
Les normes de pureté pharmaceutique s’articulent autour de deux piliers complémentaires : les monographies de la Pharmacopée Européenne et les lignes directrices ICH. Leur articulation définit le cadre réglementaire applicable à tout médicament mis sur le marché européen.
| Référentiel | Objet | Application |
|---|---|---|
| Ph. Eur. (monographies) | Spécifications par impureté nommée, seuils d’identification et de qualification | Contrôle de lot, libération, stabilité |
| ICH Q3A/Q3B | Impuretés dans les nouvelles substances actives et produits finis | Dossier d’enregistrement, specification setting |
| ICH Q3C | Solvants résiduels, classes 1 à 3 | Limites en ppm selon toxicité |
| ICH M7 | Impuretés mutagènes, évaluation TTC | Calcul de l’apport journalier acceptable |
| EDQM Module 3 | Contrôle des impuretés dans la Ph. Eur., intégration ICH | Formation, harmonisation des pratiques |
Le processus de specification setting conditionne directement la conformité des lots. Les étalons de référence et la cohérence des procédures analytiques sont cruciaux pour la robustesse des décisions qualité. Une spécification mal justifiée dans le dossier d’enregistrement peut conduire à des non-conformités lors des inspections de l’EMA ou de l’ANSM, voire à des rappels de lots.
La réglementation sur la pureté pharmaceutique impose également que chaque impureté identifiée au-delà du seuil de qualification soit caractérisée toxicologiquement. Ce n’est pas une formalité administrative. C’est une exigence scientifique qui protège directement le patient et engage la responsabilité du fabricant.
Comment la pureté influence-t-elle la sécurité et l’efficacité des médicaments ?
L’impact de la pureté sur la sécurité et l’efficacité des médicaments se manifeste à plusieurs niveaux, du mécanisme d’action moléculaire jusqu’à la gestion des risques en production industrielle.
- Efficacité thérapeutique : une teneur en principe actif inférieure aux spécifications réduit directement la dose délivrée au patient. Pour des médicaments à marge thérapeutique étroite comme la warfarine ou la digoxine, un écart de 2 à 3 % sur la pureté peut avoir des conséquences cliniques mesurables.
- Toxicité des impuretés : l’exemple du 4-aminophénol, impureté de dégradation du paracétamol, illustre le risque concret. Cette molécule est néphrotoxique à des concentrations bien inférieures à celles du principe actif. Sa présence au-delà des limites réglementaires transforme un médicament courant en produit potentiellement dangereux.
- Qualité de l’eau pharmaceutique : le contrôle qualité de l’eau pharmaceutique surveille des paramètres clés comme la conductivité et le carbone organique total (COT), indispensables pour garantir la pureté des solutions injectables et des formes liquides. Une eau hors spécification contamine l’ensemble du lot de fabrication.
- Gestion des risques et isolement de lots : un défaut de pureté détecté en cours de production est traité comme un risque majeur pouvant conduire à l’isolement de lots ou à l’arrêt de la production. Cette réponse immédiate est rendue possible par la surveillance continue via conductivité, COT et autres paramètres critiques.
Conseil de pro: Dans un contexte de recherche sur des peptides ou des réactifs reconstitués, la pureté de l’eau de reconstitution est aussi déterminante que celle du principe actif lui-même. Consultez le guide sur le contrôle qualité de l’eau bactériostatique pour comprendre les paramètres à surveiller dans votre laboratoire.
Points clés
La pureté pharmaceutique n’est pas un chiffre unique mais un profil de conformité multi-paramétrique, défini par type d’impureté et validé par des méthodes analytiques certifiées selon les référentiels Ph. Eur. et ICH.
| Point | Détails |
|---|---|
| Définition quantitative | La pureté s’exprime par %pureté = (Cexp/Ctheo) × 100, comparant concentration mesurée et théorique. |
| Spécifications par impureté | Deux lots à teneur identique peuvent avoir des conformités différentes selon les impuretés critiques présentes. |
| Méthodes analytiques validées | La HPLC avec étalonnage externe reste la référence, sous réserve de validation ICH Q2(R1) complète. |
| Cadre réglementaire | Ph. Eur., ICH Q3A/B/C et M7 définissent les seuils et procédures pour chaque catégorie d’impureté. |
| Impact sur la sécurité | Un défaut de pureté, même mineur, peut conduire à l’isolement de lots et à des risques cliniques directs. |
Ce que l’expérience terrain révèle sur le contrôle de la pureté
Après des années à observer des laboratoires de R&D et des unités de contrôle qualité, je suis convaincu que le principal angle mort dans la gestion de la pureté n’est pas technique. C’est organisationnel. Les équipes maîtrisent généralement bien la HPLC et connaissent les lignes ICH. Là où les systèmes vacillent, c’est dans la cohérence des étalons de référence utilisés d’une campagne analytique à l’autre et dans la traçabilité des décisions de specification setting.
J’ai vu des laboratoires parfaitement équipés produire des données de pureté non exploitables parce que l’étalon de référence avait été substitué sans réévaluation de la méthode. Ce type d’erreur silencieuse est plus fréquent qu’on ne le croit, et il échappe souvent aux audits internes parce qu’il ne génère pas d’alarme analytique visible.
Sur les impuretés émergentes, notamment les N-nitrosamines, le défi réel est la sensibilité analytique requise. Les seuils réglementaires se situent à des niveaux de quelques dizaines de nanogrammes par jour, ce qui impose des méthodes LC-MS/MS très spécifiques que tous les laboratoires ne maîtrisent pas encore. Les méthodes de purification des peptides et des réactifs de recherche suivent la même logique : la pureté se gagne en amont, dans la qualité des intrants, pas seulement en fin de chaîne analytique.
Mon conseil aux chercheurs universitaires : traitez le contrôle de la pureté comme un système, pas comme une série de tests isolés. La robustesse vient de la cohérence entre les étalons, les méthodes, les spécifications et la surveillance continue. Un seul maillon faible suffit à invalider l’ensemble.
— Ragnar
Herbilabs : des réactifs et solutions conçus pour la pureté analytique
Pour les chercheurs qui travaillent sur des peptides ou des réactifs reconstitués, la pureté des solutions de base conditionne la fiabilité de chaque expérience. Herbilabs fabrique ses solutions d’eau bactériostatique et ses diluants stériles selon des normes de contrôle qualité strictes, avec surveillance de la conductivité et du COT à chaque lot.
Les bénéfices des réactifs purs sur la reproductibilité des résultats sont documentés : une solution de reconstitution hors spécification peut introduire des impuretés ioniques ou organiques qui faussent vos analyses de pureté en aval. Herbilabs livre dans toute l’Europe avec documentation qualité complète. Consultez la FAQ eau bactériostatique pour comprendre comment nos produits répondent aux exigences de pureté de votre laboratoire.
FAQ
Qu’est-ce que la pureté pharmaceutique exactement ?
La pureté pharmaceutique désigne le rapport entre la concentration réelle d’une substance active mesurée analytiquement et sa concentration théorique attendue, exprimé en pourcentage. Elle se définit également par un profil de conformité par type d’impureté selon les monographies de la Ph. Eur. et les lignes ICH.
Quelle méthode analytique est utilisée pour mesurer la pureté ?
La HPLC par étalonnage externe est la méthode de référence pour l’analyse de la pureté des principes actifs. Elle doit être validée selon ICH Q2(R1) pour garantir spécificité, linéarité et exactitude avant toute utilisation en contrôle de lot.
Quelles sont les principales normes réglementaires sur la pureté pharmaceutique ?
Les normes de pureté pharmaceutique reposent sur la Pharmacopée Européenne (Ph. Eur.), les lignes ICH Q3A, Q3B, Q3C et M7, ainsi que sur les modules de formation EDQM. Ces référentiels définissent les seuils d’identification, de qualification et de limite pour chaque catégorie d’impureté.
Pourquoi deux lots peuvent-ils avoir la même teneur mais des conformités différentes ?
La conformité dépend non seulement de la teneur en principe actif, mais aussi du profil qualitatif des impuretés présentes. Un lot contenant une impureté mutagène au-delà du seuil ICH M7 sera non conforme même si son pourcentage de pureté global est identique à celui d’un lot conforme.
Comment la pureté de l’eau affecte-t-elle la qualité pharmaceutique ?
L’eau pharmaceutique hors spécification introduit des impuretés ioniques et organiques dans les formulations, compromettant directement la pureté du produit fini. Sa surveillance continue par conductivité et carbone organique total est une exigence réglementaire qui déclenche des actions qualité immédiates en cas d’anomalie.


