Private Label, White Label, Wholesale partnerships available - EU, USA and UK - Free shipping from €75
Sterile Wasserherstellung: Methoden, Sicherheit, Labor
Sterile Wasserherstellung für Forscher: Methoden, Validierung, Lagerung und typische Fehlerquellen bei der WFI-Herstellung für Peptidforschung und Labor.
TL;DR:
- Echtes sterile Wasser für Injektionen erfüllt strenge Normen gegen Endotoxine und Mikroben.
- Destillation gilt als Goldstandard, kalte Verfahren sind energieeffizienter, erfordern aber strenges Monitoring.
- Kontinuierliche Zirkulation und richtige Lagerung verhindern Biofilme und Kontaminationen im Wasser.
Wer im Labor mit Peptiden arbeitet, kennt das Problem: Das Wasser sieht klar aus, riecht neutral und scheint einwandfrei. Trotzdem kann es die Forschungsergebnisse ruinieren. Steriles Wasser ist nämlich nicht gleich Wasser für Injektionen (WFI), und dieser Unterschied ist entscheidend. Die Herstellung von wirklich sterilem Wasser für Forschungszwecke folgt strengen Normen, mehreren Produktionsschritten und komplexen Validierungsanforderungen. Viele Forscher unterschätzen, wie viele Fehlerquellen auf dem Weg zur echten Sterilität lauern. Dieser Artikel zeigt, welche Methoden es gibt, worauf bei Lagerung und Validierung zu achten ist und warum Qualität bei der Wasserherstellung keine Kompromisse erlaubt.
Inhaltsverzeichnis
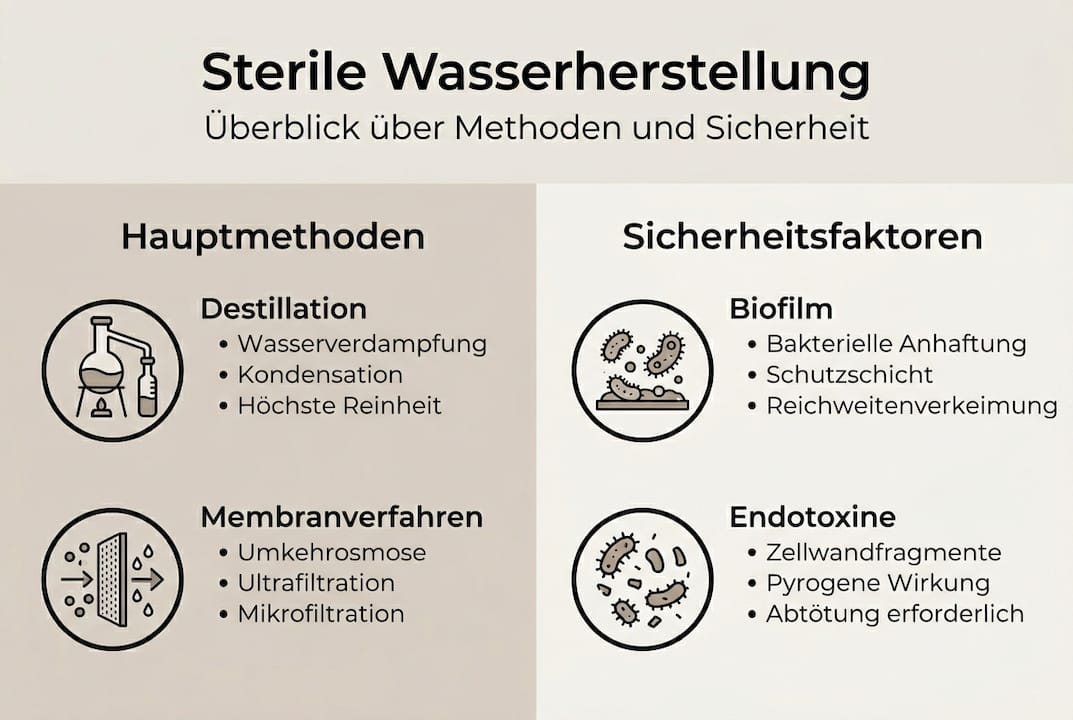
- Definition und Anforderungen von sterilem Wasser
- Hauptmethoden zur Herstellung von sterilem Wasser
- Vorbehandlung, Lagerung und Distribution
- Validierung und typische Fehlerquellen
- Unsere Sicht: Worauf es wirklich bei steriler Wasserherstellung ankommt
- Weiterführende Lösungen und Produkte für Ihr Labor
- Häufig gestellte Fragen zur sterilen Wasserherstellung
Wichtige Erkenntnisse
| Punkt | Details |
|---|---|
| Strenge Qualität unverzichtbar | Nur ultra-reines, apyrogenes Wasser ist für Forschung und Reconstitution wirklich sicher. |
| Verfahren mit Vor- und Nachteilen | Destillation ist robust, kalte Filtration energieeffizient – beide erfordern spezifische Überwachung. |
| Korrekter Umgang verhindert Biofilm | Fortlaufende Zirkulation und Sanitisierung schützen vor Biofilm und Kontamination. |
| Validierung als Erfolgsfaktor | Regelmäßige Tests und Validierungsphasen sind essenziell für sterile Wasserherstellung. |
Definition und Anforderungen von sterilem Wasser
Steriles Wasser klingt nach einem einfachen Konzept. In der Praxis steckt dahinter ein komplexes Regelwerk, das zwischen verschiedenen Wasserqualitäten klar unterscheidet. Sterile Wasserherstellung bezieht sich primär auf die Produktion von Wasser für Injektionszwecke (WFI), das ultrarein, apyrogen und für parenterale Anwendungen geeignet ist. Das bedeutet: kein einfaches abgekochtes Wasser, kein gefiltertes Leitungswasser.
WFI muss mehrere strenge Kriterien gleichzeitig erfüllen. Es geht nicht nur um Keimfreiheit, sondern auch um Apyrogenität, also die Abwesenheit von Pyrogenen wie bakteriellen Endotoxinen, die selbst nach dem Abtöten von Bakterien im Wasser verbleiben können. Zusätzlich müssen Partikelfreiheit, ein extrem niedriger Leitfähigkeitswert und ein kontrollierter pH-Wert gewährleistet sein. Die Qualität von Injektionswasser ist damit weit anspruchsvoller als bei Standard-Laborwasser.
Die wichtigsten Kriterien für WFI laut Europäischem Arzneibuch (EP) und anderen Pharmakopöen:
- Endotoxin-Grenzwert: maximal 0,25 IE/ml
- Leitfähigkeit: unter 1,3 µS/cm bei 25°C
- TOC (Gesamter organischer Kohlenstoff): maximal 500 µg/l
- Mikrobiologische Reinheit: maximal 10 KBE/100 ml
- Partikelfreiheit: strenge Grenzwerte für unlösliche Partikel
Einfaches steriles Wasser, etwa aus einer Apotheke oder einem Standardlabor, erfüllt diese Anforderungen oft nicht vollständig. Es kann zwar steril im mikrobiologischen Sinne sein, aber Endotoxine oder organische Rückstände enthalten. Für die Anwendung steriler Reagenzien in der Peptidforschung ist das ein ernstes Problem.
Wichtig: Apyrogenität ist nicht dasselbe wie Sterilität. Ein Wasser kann steril sein und trotzdem Endotoxine enthalten, die biologische Assays verfälschen oder Entzündungsreaktionen auslösen.
Die Regulierung erfolgt in Europa primär über das Europäische Arzneibuch (EP), das seit 2017 auch membranbasierte Herstellungsverfahren als gleichwertig zur Destillation anerkennt. Für Forscher bedeutet das: Nur Wasser, das nach diesen Normen produziert und dokumentiert wurde, ist für anspruchsvolle Laboranwendungen wirklich geeignet.
Hauptmethoden zur Herstellung von sterilem Wasser
Die Wahl der Herstellungsmethode bestimmt maßgeblich die Qualität und Zuverlässigkeit des Endprodukts. Traditionell setzt die Industrie auf Destillation, insbesondere Multi-Effekt-Destillation und Dampfkompression. Seit der EP-Revision 2017 sind auch kalte Verfahren wie Umkehrosmose (RO) kombiniert mit Ultrafiltration (UF) und steriler Filtration (0,1 bis 0,2 µm) offiziell anerkannt.
Hier ein direkter Vergleich der wichtigsten Methoden:
| Methode | Energiebedarf | Pyrogen-Sicherheit | Investitionskosten | Eignung für Peptide |
|---|---|---|---|---|
| Multi-Effekt-Destillation | Hoch | Sehr hoch | Hoch | Sehr gut |
| Dampfkompression | Mittel bis hoch | Sehr hoch | Mittel | Sehr gut |
| RO + UF + Sterilfiltration | Niedrig | Gut (mit Kontrolle) | Niedriger | Gut bis sehr gut |
Die Destillation gilt als Gold-Standard, weil Hitze zuverlässig Pyrogene zerstört und keine Membran versagen kann. Der Nachteil: hoher Energieverbrauch und aufwendige Infrastruktur. Für kleinere Forschungseinrichtungen ist das oft nicht wirtschaftlich.

Die kalten Verfahren sind energieeffizienter und flexibler, aber sie stellen höhere Anforderungen an das Monitoring. Eine Endotoxin-Kontamination kann bei membranbasierten Systemen auftreten, wenn Membranen altern oder Biofilm entsteht. Das erfordert konsequente Überwachung.
Die Schritte bei der kalten WFI-Herstellung im Überblick:
- Vorbehandlung: Enthärtung, Aktivkohlefilter, Partikelfilter
- Umkehrosmose (RO): Entfernung von Ionen, organischen Verbindungen, Keimen
- Ultrafiltration (UF): Entfernung von Endotoxinen und Makromolekülen
- Sterile Filtration: 0,1 bis 0,2 µm Filter als letzte Barriere
- Qualitätskontrolle: TOC, Leitfähigkeit, Endotoxin-Test
Für die Reaktivierung von Peptiden empfiehlt sich WFI aus einem validierten System, unabhängig von der Herstellungsmethode. Entscheidend ist die dokumentierte Qualität, nicht die Produktionstechnologie. Wer eigene Systeme betreibt, findet im Leitfaden zur Herstellung von Injektionswasser praktische Hinweise zur sicheren Umsetzung.
Profi-Tipp: Wenn Sie membranbasierte Systeme nutzen, prüfen Sie die Membranintegrität mindestens quartalsweise. Eine defekte UF-Membran lässt Endotoxine passieren, ohne dass dies am Aussehen des Wassers erkennbar ist. Für die Peptid-Reconstitution kann das fatale Folgen für die Reproduzierbarkeit haben.
Vorbehandlung, Lagerung und Distribution
Selbst perfekt hergestelltes WFI kann durch falsche Lagerung oder Distribution zur Kontaminationsquelle werden. Die Vorbehandlung des Rohwassers ist dabei der erste kritische Schritt. Vorbehandlungsschritte umfassen Enthärtung, Aktivkohlefilter zur Chlorentfernung und Partikelfilter, die organische Verbindungen und Schwebstoffe aus dem Rohwasser entfernen.
Ohne diese Vorbehandlung werden nachgelagerte Membranen und Filter schnell belastet und verlieren ihre Effizienz. Chlor zum Beispiel schädigt RO-Membranen irreversibel. Das ist kein theoretisches Problem, sondern ein häufiger Praxisfehler in Labors, die Systeme ohne ausreichende Vorbehandlung betreiben.
Bei der Lagerung und Distribution gibt es zwei grundlegende Ansätze:
| Methode | Temperatur | Vorteile | Risiken |
|---|---|---|---|
| Heißer Loop | >70°C | Verhindert Biofilm zuverlässig | Hoher Energiebedarf, Materialbelastung |
| Kalter Loop + UV/Chemie | Raumtemperatur | Energieeffizienter | Biofilm-Risiko ohne konsequente Sanitisierung |
Heiße Loops über 70°C gelten als zuverlässigste Methode zur Biofilm-Prävention. Kontinuierliche Zirkulation verhindert Stagnation, die Biofilm begünstigt. Kalte Systeme erfordern regelmäßige chemische oder UV-Sanitisierung als Ausgleich.
Die wichtigsten Punkte zur sicheren Lagerung:
- Keine Toträume (Dead Legs) im Leitungssystem, da sich dort Biofilm ungestört entwickeln kann
- Kontinuierliche Zirkulation im Loop, auch wenn kein Wasser entnommen wird
- Regelmäßige Sanitisierung mit Heißdampf, Ozon oder validierten Chemikalien
- Drucküberwachung und Temperaturprotokollierung an kritischen Punkten
- Verwendung von Edelstahl (316L) oder validierten Kunststoffen für alle wasserführenden Teile
Für Forscher, die auf sichere Lagerung von sterilem Wasser angewiesen sind, ist die Wahl des richtigen Systems keine optionale Entscheidung. Ein kontaminiertes Lagersystem macht alle vorherigen Produktionsschritte wertlos. Besonders in kleinen Forschungseinrichtungen wird dieser Aspekt systematisch unterschätzt.
Endotoxine sind dabei die gefährlichste unsichtbare Bedrohung. Sie entstehen durch Biofilm, überleben Sterilfiltration und lassen sich nur durch Hitze oder spezielle Depyrogenisierung zuverlässig eliminieren. Wer kalte Systeme betreibt, muss das Endotoxin-Monitoring entsprechend intensivieren.
Validierung und typische Fehlerquellen
Ein WFI-System ist erst dann einsatzbereit, wenn es vollständig validiert wurde. Die Validierung folgt den klassischen GMP-Phasen: IQ (Installationsqualifizierung), OQ (Betriebsqualifizierung) und PQ (Leistungsqualifizierung). Jede Phase hat spezifische Anforderungen und Zeitrahmen.
Der Validierungsprozess im Überblick:
- IQ (Installation Qualification): Überprüfung, ob alle Komponenten korrekt installiert und dokumentiert sind
- OQ (Operational Qualification): Nachweis, dass das System unter definierten Bedingungen korrekt funktioniert
- PQ (Performance Qualification): Beweis der dauerhaften Leistungsfähigkeit unter Realbedingungen, tägliche Probenahme für Mikrobiologie und Endotoxine über mehrere Monate
Die PQ-Phase ist dabei besonders zeitaufwendig. Tägliche Probenahmen für Mikroben und Endotoxine über Monate hinweg sind keine Übertreibung, sondern notwendig, um saisonale Schwankungen und Systemverhalten unter verschiedenen Betriebsbedingungen zu erfassen.
Kritischer Hinweis: Viele Labors beenden die intensive Überwachung nach der Validierung zu früh. Ein System, das in der PQ-Phase einwandfrei war, kann Monate später durch Membranverschleiß oder Biofilmbildung in Dead Legs zum Problem werden.
Die häufigsten Fehlerquellen in der Praxis:
- Dead Legs: Toträume im Rohrsystem, in denen Wasser stagniert und Biofilm entsteht
- Temperaturschwankungen: Ungleichmäßige Heizung im Loop ermöglicht Keimwachstum in kälteren Zonen
- Membranverschleiß: Alte oder beschädigte UF-Membranen lassen Endotoxine passieren
- Unzureichende Sanitisierung: Zu seltene oder nicht vollständig validierte Sanitisierungszyklen
- Falsche Probenahme: Nicht-repräsentative Entnahmepunkte verfälschen das Monitoring
Bei kalter WFI-Herstellung sind die Risiken durch Biofilm und Endotoxin-Kontamination besonders relevant, weil die thermische Barriere fehlt. Das erfordert eine strengere Filtration und häufigere Kontrollen als bei Destillationssystemen.
Profi-Tipp: Führen Sie einen Trend-Analyse-Plan für alle Monitoring-Daten ein. Einzelne Ausreißer sind oft weniger problematisch als schleichende Verschlechterungen über Wochen. Wer Trendlinien verfolgt, erkennt Systemprobleme, bevor Grenzwerte überschritten werden. Weitere Informationen zu Endotoxin-Überwachung und Verdünnern für die Peptidforschung finden Sie in unseren weiterführenden Ressourcen.
Unsere Sicht: Worauf es wirklich bei steriler Wasserherstellung ankommt
Nach Jahren in der Versorgung von Forschungseinrichtungen mit sterilen Produkten fällt uns ein Muster auf: Die meisten Probleme entstehen nicht bei der Herstellung, sondern danach. Systeme werden validiert, dann läuft der Alltag und das Monitoring schleifen. Destillation ist robust und verzeihend. Membranbasierte Verfahren sind effizienter, aber sie bestrafen Nachlässigkeit sofort.
Das Unbequeme daran: Endotoxin-Kontamination ist unsichtbar. Wasser kann kristallklar sein und trotzdem Peptid-Assays ruinieren. Viele Forscher entdecken das Problem erst, wenn Ergebnisse nicht reproduzierbar sind. Dann beginnt die aufwendige Fehlersuche.
Unsere klare Empfehlung: Qualität vor Geschwindigkeit. Wer auf zertifiziertes WFI aus validierten Produktionssystemen setzt, statt auf selbst hergestelltes Wasser aus unzureichend überwachten Systemen, spart langfristig Zeit und Ressourcen. Ein Vergleich zwischen bakteriostatischem und sterilem Wasser zeigt außerdem, welche Wasserart für welche Anwendung wirklich geeignet ist.
Weiterführende Lösungen und Produkte für Ihr Labor
Wenn Sie sich nach diesem Artikel fragen, wie Sie die Qualität Ihrer Reconstitution-Lösungen praktisch sicherstellen können, haben wir bei Herbilabs konkrete Antworten. Unsere Produkte werden in einer dedizierten Einrichtung nach strengen Reinheitsstandards hergestellt und sind speziell für die Anforderungen der Peptidforschung ausgelegt.
Ob Sie Fragen zu bakteriostatischem Wasser haben, einen fundierten Vergleich zwischen bakteriostatischem und sterilem Wasser suchen oder mehr über die Qualität unseres Injektionswassers erfahren möchten: Wir unterstützen Sie mit verifizierten Produkten, transparenten Qualitätsdaten und kompetenter Beratung. Forscher in ganz Europa vertrauen auf unsere zuverlässige Lieferung und gleichbleibende Produktqualität.
Häufig gestellte Fragen zur sterilen Wasserherstellung
Was ist der Unterschied zwischen sterilem Wasser und Wasser für Injektionen (WFI)?
Wasser für Injektionen ist ultrarein, apyrogen und speziell für parenterale Anwendungen validiert, während einfaches steriles Wasser diese Anforderungen nicht sicher erfüllt. WFI unterliegt strengeren Grenzwerten für Endotoxine, Leitfähigkeit und organischen Kohlenstoff.
Warum werden moderne kalte Verfahren bevorzugt, obwohl sie für Biofilm anfällig sind?
Kalte membranbasierte Verfahren wie Umkehrosmose sind energieeffizienter und kostengünstiger als Destillation, erfordern aber regelmäßige Kontrolle und konsequentes Monitoring zum Schutz vor Biofilm und Endotoxinen. Der Vorteil liegt im geringeren Betriebsaufwand bei ausreichend überwachten Systemen.
Wie kann Biofilm und Endotoxinbildung in WFI-Systemen verhindert werden?
Durch kontinuierliche Zirkulation und heiße Loops über 70°C sowie regelmäßige chemische oder UV-Sanitisierung lässt sich Biofilmbildung effektiv vermeiden. Dead Legs im Rohrsystem müssen konstruktiv eliminiert werden.
Welche Validierungsphasen sind bei der Herstellung von sterilem Wasser notwendig?
Die Validierung umfasst IQ, OQ und PQ mit täglicher Mikrobiologie und Endotoxin-Kontrolle; diese Prozesse dauern mehrere Monate und müssen vollständig dokumentiert werden, bevor ein System in Betrieb genommen wird.


