Private Label, White Label, Wholesale partnerships available - EU, USA and UK - Free shipping from €75
GMP labware standards for reliable bacteriostatic water
Discover what GMP manufacturing in labware means for ensuring bacteriostatic water purity. Learn essential compliance standards today!
TL;DR:
- Not all sterile labware meets GMP standards, which are essential for ensuring contaminant control and process validation. GMP involves documented, validated procedures, rigorous environmental controls, and traceability to prevent silent failure modes that compromise research integrity. Proper material selection, cleaning validation, and thorough documentation are critical to compliant manufacturing of bacteriostatic water and other sensitive solutions.
Not all sterile labware is GMP-compliant, and that distinction matters far more than most labs initially realize. A vial labeled “sterile” may pass a basic contamination check yet still leach trace metals, harbor residual detergent, or lack validated container closure integrity, each of which can silently compromise bacteriostatic water purity and destabilize research outcomes. Good Manufacturing Practice (GMP) sets a far more demanding bar: documented processes, validated cleaning, material-specific standards, and regulatory traceability built into every production step. This article unpacks exactly what GMP means for labware manufacturing, how it applies to bacteriostatic water specifically, and what compliance looks like in practice for European and UK research facilities.
Table of Contents
- Understanding GMP: What it means for labware manufacturing
- GMP labware requirements for bacteriostatic water
- Labware cleaning, validation, and common edge cases
- Verification, documentation, and regulatory alignment in Europe and the UK
- The hidden challenges most labs overlook in GMP labware standards
- Find reliable GMP labware for bacteriostatic water and more
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| GMP demands more than sterility | Labware for bacteriostatic water must meet strict GMP standards far beyond typical ‘sterile’ labels—including rigorous documentation and validation. |
| Material selection is critical | Borosilicate, quartz, and PFA are chosen for their low trace metals and chemical stability in critical applications. |
| Cleaning and verification are core | Automated cleaning and validated testing are required to meet purity and integrity benchmarks under GMP guidelines. |
| Documentation proves compliance | Audit-ready records of cleaning, sterility, and environmental control are essential for regulatory approval. |
| Shortcuts risk research and safety | Neglecting GMP labware standards can compromise results and put labs at risk of regulatory action. |
Understanding GMP: What it means for labware manufacturing
GMP, short for Good Manufacturing Practice, is a regulatory framework that governs how products are manufactured, tested, and controlled to ensure consistent quality. For labware, the regulatory landscape in Europe and the UK is defined by several overlapping frameworks. EudraLex Volume 4 covers GMP for medicinal products, the MHRA enforces these standards within the UK post-Brexit, and ISO 13485 applies specifically to medical device manufacturing. Together, they demand dedicated production areas, HVAC-controlled environments, and thoroughly validated processes.
For labware intended to hold or process products like bacteriostatic water, these requirements go well beyond the ordinary. Bacteriostatic water contains 0.9% benzyl alcohol as a preservative, making it sensitive to leachable contaminants that could react with or degrade the preservative system. Any labware failure, whether a compromised seal or a contaminated surface, feeds directly into the solution.
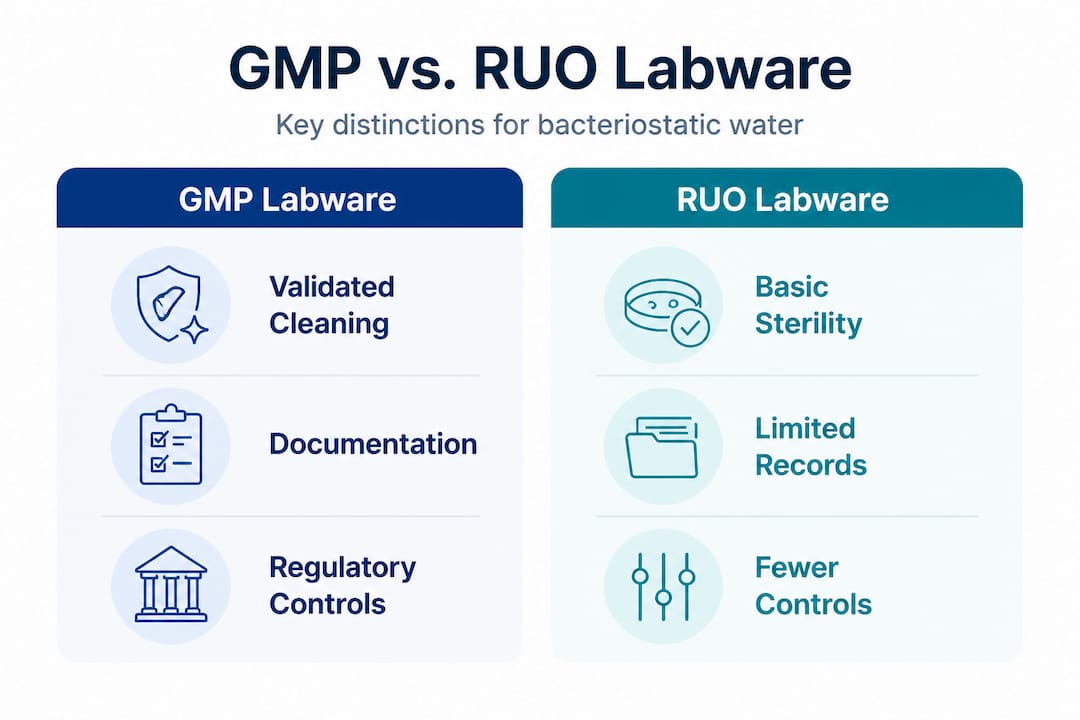
A critical distinction that many researchers overlook is the difference between RUO (Research Use Only) and GMP-grade labware:
- RUO labware is designed for general laboratory research without regulatory oversight or mandated validation.
- GMP labware requires documented environmental controls, validated cleaning procedures, traceability records, and material qualification data.
- RUO-to-GMP transition demands enhanced documentation at every stage, including batch records, deviation logs, and environmental monitoring reports.
- Cross-contamination controls under GMP mandate physically separated production areas, HEPA-filtered air handling, and pressure cascades to protect product integrity.
- Validated processes must be reproducible across batches, not just demonstrated once, meaning every manufacturing run generates evidence of compliance.
“Preventing cross-contamination via dedicated areas, HVAC controls, and validated processes is a foundational requirement under EudraLex Vol 4 GMP and MHRA guidelines, not an optional quality add-on.”
The practical difference is significant. An RUO vial might be perfectly adequate for a routine reagent storage application. Put that same vial into a production line for injectable-grade bacteriostatic water, and you are operating outside validated parameters. The consequences range from failed quality control batches to regulatory non-compliance and, most critically, compromised research reproducibility.
GMP labware requirements for bacteriostatic water
With GMP principles defined, let’s map the specific requirements that make labware appropriate for sensitive solutions like bacteriostatic water. These requirements span material selection, sterility benchmarks, and physical integrity, and they are not arbitrary. Each standard connects directly to a known failure mode.
Step-by-step GMP labware requirements:
-
Material qualification: Primary container materials must be chemically inert under expected conditions. Borosilicate glass Type I (HGB 1 hydrolytic resistance) is the standard choice for vials, paired with butyl rubber stoppers that minimize extractables. For applications requiring sub-ppt (parts per trillion) metal purity, quartz or perfluoroalkoxy (PFA) polymer labware replaces borosilicate entirely.
-
Leachables and extractables control: GMP labware must minimize leachable trace metals to below ppt levels and account for nanoplastic shedding from polymer components. These thresholds matter because even low-level metal contamination can catalyze benzyl alcohol degradation in bacteriostatic water.
-
Sterility validation: All primary labware for bacteriostatic water production must meet USP <71> sterility testing requirements. This is not a one-time test but an ongoing validation activity tied to each production batch.
-
Endotoxin testing: Bacterial endotoxins must remain below 0.25 EU/mL per USP <85>. Endotoxin contamination can survive standard sterilization and is one of the most insidious quality failures in injectable solutions.
-
Container closure integrity: Every vial seal must be validated through microbial ingress testing and seal strength measurement. A closure that physically holds may still allow microbiological breach under certain conditions, which is why both mechanical and biological validation methods are required.
| Labware property | Standard/requirement | Test method |
|---|---|---|
| Hydrolytic resistance | Type I HGB 1 | Ph. Eur. 3.2.1 |
| Endotoxin limit | <0.25 EU/mL | USP <85> LAL test |
| Sterility | No growth in 14-day incubation | USP <71> |
| Extractable metals | Sub-ppt levels | ICP-MS analysis |
| Closure integrity | No microbial ingress | Microbial challenge test |
| Nanoplastic shedding | Below validated threshold | Laser diffraction |
Pro Tip: When evaluating a labware supplier, ask specifically for their extractable/leachable (E&L) study data for the container-closure system you intend to use. Suppliers who cannot provide batch-specific E&L documentation are not operating under full quality assurance principles, regardless of any GMP label they apply to their products.
The distinction between sterile and GMP-validated is perhaps sharpest here. Sterility tells you that viable microorganisms were absent at the point of testing. GMP validation tells you why they were absent, how you know the process that produced that result is reproducible, and what documentation exists to prove it six months from now during an audit.
Labware cleaning, validation, and common edge cases
Even the most carefully selected materials fail without rigorous, validated cleaning. Cleaning validation is one of the most technically demanding aspects of GMP labware compliance, and it is also where many labs, particularly those transitioning from RUO practices, encounter the most problems.

Manual versus automated cleaning:
Manual cleaning introduces inherent variability. Technician technique, water temperature, detergent concentration, and rinse duration all fluctuate between operators and shifts. In contrast, automated washers deliver reproducible cycles with controlled parameters, validated cycle times, and documented outputs. For GMP compliance, automated cleaning is not just preferred. For applications demanding sub-ppt metal levels, it is the only defensible choice.
Common residue risks:
- Phosphate-based detergents are a frequent source of assay interference, particularly in enzyme-linked assays where phosphate ions affect substrate turnover rates.
- Surfactant residues lower surface tension unpredictably, which can affect critical filling operations for bacteriostatic water vials.
- Acid leaching, used to remove trace metals from borosilicate surfaces, must itself be validated and performed with automated equipment to ensure consistency.
Critical benchmarks for cleaning validation:
| Parameter | GMP benchmark | Measurement method |
|---|---|---|
| Residue removal | >99% removal demonstrated | TOC and conductivity testing |
| Surface roughness | ≤0.8 μm Ra | Profilometry |
| Dead leg geometry | Ratio below 3D (diameter) | Piping/vessel design review |
| Rinse water quality | TOC <500 ppb, conductivity <1.3 μS/cm | Online TOC monitor |
Dead legs deserve specific attention. A dead leg is a section of tubing or vessel where liquid stagnates rather than flowing freely. In water storage and handling systems, dead legs with a ratio above 3D create conditions for biofilm formation, even in systems that appear clean. The physical design of labware and its associated handling equipment is therefore a GMP variable, not just an engineering detail.
When are quartz and PFA labware necessary? For trace metal work requiring ppt-level cleanliness, borosilicate glass simply cannot meet the specification, even after acid leaching. Quartz has far lower metallic impurity content in the glass matrix itself, and PFA offers both chemical inertness and minimal ion exchange with aqueous solutions. These materials are also essential when working with lab water quality standards that demand sub-ppb elemental baselines.
Pro Tip: Always verify rinse water quality at the point of final rinse, not just at the inlet. Water quality degrades as it passes through tubing and vessel surfaces, and measuring only at the source can give a false picture of cleanliness at the labware surface.
Verification, documentation, and regulatory alignment in Europe and the UK
Cleaning and process validation are only credible when documented and verified. For European and UK labs, regulatory alignment means producing audit-ready records that satisfy both the MHRA and EudraLex Vol 4 requirements simultaneously.
What verification requires:
-
Hydrolytic and acid resistance testing: Borosilicate Type I glass must demonstrate HGB 1 hydrolytic resistance and S1 acid resistance per Ph. Eur. 3.2.1 before it qualifies for bacteriostatic water contact.
-
Cleaning validation documentation: Every cleaning cycle must be validated to show >99% residue removal, with TOC and conductivity as the primary analytical endpoints. Validation must cover worst-case conditions, including maximum soiling, minimum detergent concentration, and shortest cycle time.
-
Container integrity testing: For sterile liquid products, 100% integrity testing on all fusion-sealed containers (ampoules, for instance) is standard. This typically uses bubble or high-voltage leak detection to confirm seal integrity without requiring destructive sampling.
-
Environmental monitoring: Cleanroom and controlled-environment logs must document viable and non-viable particulate counts, temperature, humidity, and pressure differentials. These records form the environmental context that supports every batch record.
-
Batch documentation and traceability: Each production lot must be traceable through raw material certificates, in-process testing data, cleaning records, and final release testing. No single record stands alone under GMP.
Key regulatory callout: Under EudraLex Vol 4 and MHRA guidance, moving labware from RUO to GMP classification requires not just physical upgrades but a complete documentation architecture. Gaps in environmental monitoring or incomplete batch records are the most common citations during regulatory inspections.
A useful metric to benchmark compliance: water used in GMP cleaning cycles must meet USP Purified Water specifications (conductivity <1.3 μS/cm, TOC <500 ppb) as a minimum, with Water for Injection (WFI) required for final rinse steps in sterile product manufacturing. This distinction alone separates compliant from non-compliant facilities, and it is auditable from day one.
The hidden challenges most labs overlook in GMP labware standards
There is a version of GMP compliance that looks correct on paper but fails at the bench. We have seen it repeatedly, and it almost always traces back to the same three oversights.
The first is treating documentation as a retrospective activity. Teams run a cleaning validation study, write it up once, and file it away. Six months later, a technician changes the detergent brand, the cycle temperature drifts by four degrees, or a new batch of vials comes from a different sub-supplier. None of these changes trigger a formal review. The documentation no longer reflects reality. This is not hypothetical. It is the most common finding during audits, and it undermines an otherwise technically sound operation.
The second oversight is underestimating last-mile contamination. A vial can pass every validation test at the point of manufacture and then pick up contamination during transfer, storage, or secondary packaging. Particulates from carton materials, volatile organic compounds from packaging adhesives, and even static charge attracting airborne particles are all documented contamination pathways. GMP thinking requires you to treat the entire supply chain as part of the validated system, not just the production floor.
The third is the cost calculation problem. Shortcuts in cleaning validation or contamination control look cheap until a batch fails or, worse, until data from a six-month study is called into question because the primary container cannot be shown to have met validated purity standards. The cost of a failed study, or a regulatory citation, always exceeds the cost of doing it right the first time.
The practical takeaway here is that GMP compliance is an ongoing operational discipline, not a status you achieve once and maintain passively. Cleaning verification, environmental monitoring, and documentation reviews need to be integrated into routine lab management, not treated as one-off qualification activities.
Find reliable GMP labware for bacteriostatic water and more
Understanding GMP labware standards is the foundation. Sourcing from a manufacturer who applies them consistently is the next step.
At Herbilabs, our bacteriostatic water and sterile solutions are produced in a dedicated facility to strict GMP-aligned purity standards. Every product batch is supported by documented quality controls, material certificates, and endotoxin testing, so your research starts on a verified baseline. Whether you want to review our complete guide to bacteriostatic water for reference, explore answers in our bacteriostatic water FAQs, or go straight to ordering, our shop carries GMP-aligned solutions ready for UK and European delivery with wholesale pricing available for institutional buyers and resellers.
Frequently asked questions
What are the key differences between GMP and RUO labware?
GMP labware requires enhanced documentation and environmental controls, along with formally validated processes, while RUO labware is designed for research use and carries no equivalent regulatory manufacturing requirements.
Which materials are preferred for labware in bacteriostatic water production?
Borosilicate vials with butyl stoppers are the baseline standard, minimizing leachables and extractables. For applications requiring sub-ppt metal purity, quartz or PFA labware is the appropriate upgrade.
How is sterility validated in GMP labware for injectable solutions?
Sterility is validated through USP <71> sterility testing alongside container closure integrity verification, which combines microbial ingress testing with physical seal strength measurement across validated production conditions.
Why is automated labware cleaning preferred in GMP processes?
Automated washers ensure reproducibility across cycles, eliminate technician variability, and prevent detergent or phosphate residues from interfering with sensitive assays in high-purity applications.
What documentation is required to prove GMP compliance for labware?
You need cleaning validation records demonstrating >99% residue removal, environmental monitoring logs, batch records with full traceability, and 100% integrity testing data for all sterile container closures.
Recommended
- Bacteriostatic vs Sterile Water: Safe Lab Application Guide 2025 – Herbilabs Labware
- How to Store Bacteriostatic Water Safely in the Lab – Herbilabs Labware
- What is Bacteriostatic Water? Best 2025 Guide for Labs in Europe – Herbilabs Labware
- Blog – Herbilabs Labware
- Bacteriostatic water: Essential guide for peptide research


