Private Label, White Label, Wholesale partnerships available - EU, USA and UK - Free shipping from €75
Why Aseptic Processing Matters for Pharmaceutical Labs
Discover why aseptic processing matters for pharmaceutical labs. Learn how it protects product integrity and patient safety in modern medicine.
TL;DR:
- Aseptic processing is vital for preventing microbial contamination during pharmaceutical and food manufacturing, ensuring product safety and shelf stability. It involves sterilizing components separately and assembling them in a controlled sterile environment, especially crucial for biologics that cannot withstand terminal sterilization. Advances like dry aseptic systems and automation are enhancing reliability, sustainability, and regulatory compliance in this critical manufacturing process.
A single microbial contaminant can invalidate an entire pharmaceutical batch, trigger a regulatory recall, or compromise a patient’s safety. That’s why aseptic processing matters far beyond clean rooms and sterile gowns. It’s the backbone of how modern medicine and shelf-stable food reach consumers without causing harm. For food scientists, pharmaceutical researchers, and quality control professionals, understanding its mechanics and limitations is not optional. It’s the foundation of every reliable product you manufacture, test, or release.
Table of Contents
- Key takeaways
- Why aseptic processing matters: core principles and mechanisms
- Key benefits of aseptic processing for product quality and safety
- Critical control points and where aseptic systems fail
- Innovations and sustainability trends in aseptic technology
- Practical applications across labs and manufacturing
- My take on the future of aseptic investment
- Research-grade sterile solutions from Herbilabs
- FAQ
Key takeaways
| Point | Details |
|---|---|
| Aseptic processing prevents contamination | Maintaining sterile conditions throughout production protects product integrity and patient safety. |
| UHT and sterile filling work together | Food applications combine high-temperature sterilization with sterile filling to achieve 12-18 month ambient shelf life. |
| Endotoxin control requires cleaning validation | Sterilization alone doesn’t eliminate heat-stable endotoxins; validated cleaning protocols are mandatory. |
| Dry aseptic tech reduces environmental impact | Electron beam systems eliminate water and chemical rinsing, cutting waste and residual contamination risk. |
| Biologics demand aseptic methods specifically | Temperature-sensitive biologics cannot survive terminal sterilization, making aseptic fill-finish the only viable option. |
Why aseptic processing matters: core principles and mechanisms
What is aseptic processing, exactly? At its core, it is the practice of manufacturing, filling, and packaging products under conditions that prevent any microbial or particulate contamination from entering the product. Unlike terminal sterilization, where the final sealed product is sterilized after filling, aseptic processing sterilizes each component separately, then assembles everything in a controlled sterile environment.
In food manufacturing, the dominant approach is Ultra-High Temperature (UHT) processing. Product is heated to 130-150°C for 2-5 seconds, which destroys microorganisms while preserving flavor and nutritional content far better than retort processing. The product is then rapidly cooled and transferred into a pre-sterilized container within a sterile filling zone, all without exposure to ambient air.
In pharmaceutical manufacturing, the process is more complex. Liquid drug products are typically sterilized by filtration through 0.22-micron membranes, then filled into pre-sterilized vials, syringes, or cartridges inside ISO 5 (Grade A) cleanrooms with unidirectional airflow. The key difference from retort or hot-fill methods is that the drug itself is never exposed to the heat levels required for terminal sterilization. That matters enormously for biologics and complex molecules.
The essential components of an aseptic system
Every effective aseptic system relies on four interdependent elements working in coordination:
- Sterilized product or drug substance prepared through heat, filtration, or radiation before filling
- Sterilized primary packaging including vials, closures, and foils treated with steam, dry heat, or chemical agents
- Controlled filling environment with HEPA-filtered unidirectional airflow, pressure differentials, and barrier or isolator technology
- Validated cleaning and sterilization procedures for all equipment surfaces contacting the product
Pro Tip: Don’t conflate “sterile” with “endotoxin-free.” Endotoxins are heat-stable lipopolysaccharides from gram-negative bacteria. They survive standard autoclaving. Your cleaning validation program must specifically address endotoxin removal, not just bioburden reduction.
The shift from open cleanrooms to restricted access barrier systems (RABS) and pharmaceutical isolators has transformed how these environments are controlled. Isolators physically separate operators from the sterile zone, reducing the single greatest contamination vector in any aseptic operation: human activity.

Key benefits of aseptic processing for product quality and safety
The practical advantages of aseptic techniques are substantial, and they compound across product categories. The case for adoption goes well beyond regulatory pressure.
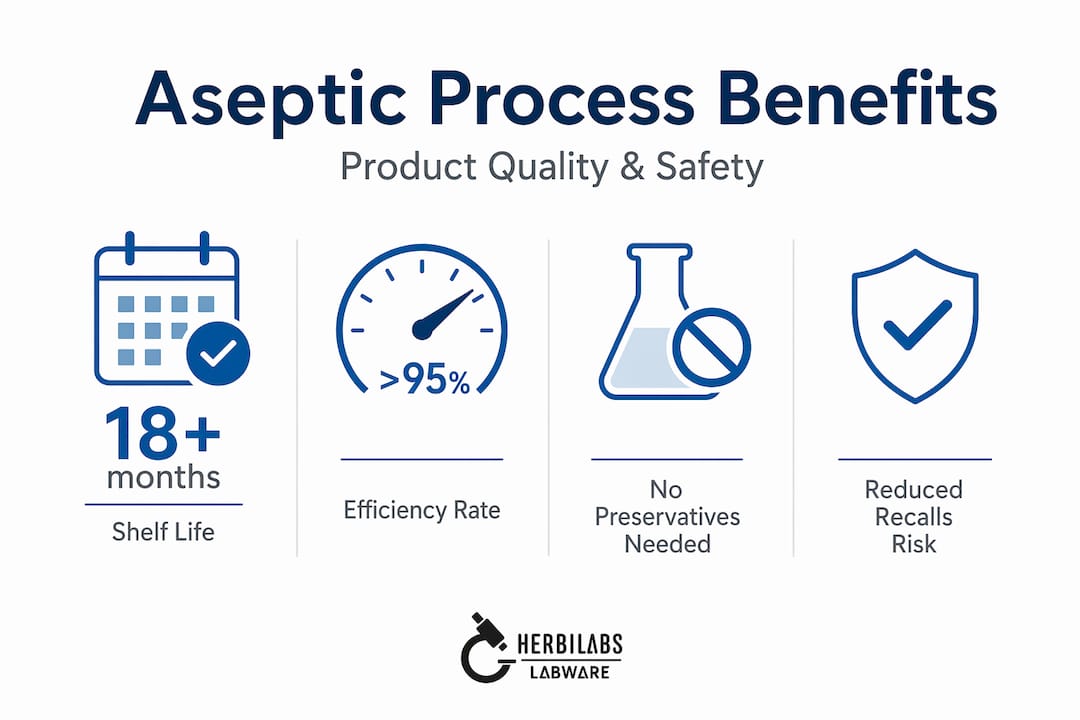
Aseptically filled products routinely achieve 12-18 months of ambient shelf life without refrigeration or preservatives. For food manufacturers, this opens global distribution channels that cold-chain-dependent products cannot access. For pharmaceutical companies, it allows room-temperature storage of injectables that would otherwise require cold-chain logistics at significant cost.
Quality preservation is the second major advantage. Because aseptic processing avoids the high sustained temperatures of retort or autoclave sterilization, it protects heat-labile compounds including vitamins, amino acids, peptides, and active pharmaceutical ingredients. In food, this translates to flavor and color profiles that are noticeably closer to fresh products. In pharma, it means potency and structural integrity for sensitive biologics and small molecules that degrade rapidly under thermal stress.
The benefits extend to compliance and risk management:
- Reduced contamination risk translates directly to fewer out-of-specification results and lower batch rejection rates
- Regulatory alignment with FDA 21 CFR Part 211, EU GMP Annex 1, and WHO guidelines on sterile manufacturing becomes more achievable when processes are designed with aseptic principles from the start
- Product recall reduction is a measurable outcome. Contaminated sterile products carry the highest recall severity classifications, and recalls cost manufacturers millions beyond the direct product loss
- Sustainability gains come from eliminating water and chemicals in dry aseptic systems, cutting waste streams and supporting corporate environmental commitments
- Market access for ambient products increases because storage and distribution infrastructure requirements drop significantly
Modern aseptic filling lines achieve mechanical efficiency rates above 95% and can run 200 or more hours continuously before requiring cleaning or sterilization cycles. That operational reliability directly affects cost-per-unit and throughput.
Critical control points and where aseptic systems fail
Understanding the importance of aseptic processing also means understanding where it breaks down. The failure modes are specific and often underappreciated by teams that focus exclusively on sterilization validation.
-
Aseptic transfer zones are the most vulnerable points in any aseptic system. Every time product, equipment, or materials cross a sterile boundary, sterility is at risk. Physical transitions during sterilization require precise control of pressure differentials and vacuum modulation to prevent filter damage and maintain barrier integrity.
-
Endotoxin contamination is a separate failure mode from microbial contamination. Endotoxins are heat-stable and require validated cleaning procedures for removal. Sterilization alone will not address them. Cleaning validation beyond sterilization is mandatory in any compliant aseptic operation.
-
Personnel behavior remains the most statistically significant source of contamination in traditional open cleanrooms. Gowning qualification, aseptic technique training, and media fill validation all exist specifically to quantify and reduce this risk.
-
Environmental monitoring gaps create blind spots. Continuous viable and non-viable particle monitoring, surface sampling, and compressed gas testing are not optional. They are the early warning system for contamination events before product is affected.
-
Equipment cleaning and passivation failures allow biofilm formation and product carryover between batches. This is where automated clean-in-place (CIP) and steam-in-place (SIP) systems have to be validated to the same rigor as the filling process itself.
Pro Tip: When auditing an aseptic system, look at personnel environmental monitoring data over time, not just the most recent cycle. Trends in personnel contamination rates reveal training gaps and behavioral drift that point-in-time audits miss entirely.
The complexity of managing all these control points simultaneously explains why skilled personnel and automation are not competing priorities but complementary ones. Closed-system sampling and real-time digital environmental monitoring reduce the human touchpoints that introduce risk.
Innovations and sustainability trends in aseptic technology
The aseptic processing sector is evolving rapidly. The global aseptic pharmaceutical processing equipment market is projected to grow from $8.58 billion in 2022 to $16.39 billion by 2031, a CAGR of 7.6%. The growth is not just volume. It reflects a fundamental shift in what aseptic systems are expected to do.
Dry aseptic technologies represent the most significant operational change in recent years. By eliminating water rinsing and chemical sterilants entirely, these systems comply with FDA residual peroxide limits of under 0.5 ppm while reducing environmental footprint. Electron beam (EB) sterilization for packaging materials and closures is replacing hydrogen peroxide-based systems in high-volume lines, with measurable reductions in chemical waste streams.
| Technology | Traditional approach | Current innovation |
|---|---|---|
| Container sterilization | Hydrogen peroxide vapor rinse | Electron beam, no chemicals or water |
| Environmental monitoring | Periodic manual plate counts | Continuous automated real-time particle monitoring |
| Sterile boundary design | Open cleanroom with RABS | Closed pharmaceutical isolators |
| Sampling during production | Manual, breaks sterility | Automated closed-system sampling |
| Sterilization validation | Periodic biological indicators | Continuous parametric release |
“Aseptic processing now encompasses sustainability and auditability alongside sterility, addressing regulatory, consumer, and corporate environmental expectations simultaneously.” (Sustainable Decontamination Solutions for Next-Gen Aseptic Lines)
Automated closed-system sampling has also redefined how manufacturers interact with running processes. Rather than stopping a line to pull a sample manually, closed sampling allows in-process intelligence without breaking sterile boundaries. For complex biologics manufacturing where every intervention carries contamination risk, this shift is operationally significant.
The rise of biologic therapies is perhaps the strongest single driver of aseptic market expansion. These products cannot tolerate terminal sterilization. Aseptic fill-finish is not one option among several. It is the only method.
Practical applications across labs and manufacturing
The why use aseptic methods question becomes concrete when you map it to specific workflows:
- Biologics and advanced therapy fill-finish: Temperature-sensitive biologics including monoclonal antibodies, cell therapies, and gene therapy vectors require aseptic processing at every stage from drug substance handling through final container filling
- Food and beverage ambient products: Juices, dairy alternatives, nutritional supplements, and soups use aseptic filling to achieve 12-18 month shelf life with quality that conventional retort processing cannot match
- Quality control laboratories: Sterile sampling, microbiological testing, and reagent preparation all depend on aseptic technique to produce valid, uncontaminated results
- Research and development labs: Peptide reconstitution, cell culture media preparation, and injectable formulation work require sterile lab techniques that apply aseptic principles at bench scale
- Contract development and manufacturing organizations (CDMOs): Aseptic processing compliance to FDA 21 CFR Part 211, EU GMP Annex 1, and WHO TRS standards is a prerequisite for any sterile contract manufacturing relationship
For quality control professionals, the intersection of lab compliance standards and aseptic practice is where documentation, validation, and operational discipline converge. Compliance with regulatory frameworks is not separate from aseptic processing. It is built into it.
My take on the future of aseptic investment
I’ve spent years working with labs and manufacturing teams that treat aseptic processing as a compliance box to check rather than a strategic manufacturing capability. That view is expensive, and it’s becoming harder to sustain.
What I’ve learned from observing facilities across pharma and food is that the gap between teams who understand aseptic principles deeply and those who follow procedures without understanding them shows up in data. It shows in environmental monitoring trends, in media fill failure rates, and in the frequency of manufacturing deviations. Procedures protect you until they don’t. Understanding protects you always.
The regulatory trajectory is clear. EU GMP Annex 1 in its revised form places greater emphasis on contamination control strategy as a holistic documented framework, not just a collection of SOPs. FDA continues to increase scrutiny on data integrity in environmental monitoring programs. The expectation is that aseptic processing facilities understand not just what they do, but why it works and what would cause it to fail.
My genuine concern is that as automation advances and closed systems reduce direct human engagement with sterile operations, some teams will mistake process reliability for process understanding. The equipment can be right while the thinking is wrong. Invest in the people as aggressively as you invest in the technology. Validated isolators and real-time monitoring are only as reliable as the people interpreting the data they produce.
— Ragnar
Research-grade sterile solutions from Herbilabs
If your work depends on aseptic handling, the quality of your sterile reagents is not a minor detail. It’s the starting point.
Herbilabs supplies bacteriostatic water, sterile diluents, and reconstitution solutions manufactured to strict purity standards in a dedicated facility. Every product in the catalog is designed for researchers who cannot afford contamination events at the reagent level. Whether you’re reconstituting peptides, preparing injectable formulations, or maintaining sterile sample integrity in a quality control lab, the foundational answers are in the bacteriostatic water FAQ resource. For guidance on choosing between sterile solution types, the bacteriostatic vs. sterile water guide covers the practical distinctions that matter in research applications. Herbilabs serves research institutions, universities, and independent researchers across the UK and Europe with reliable, research-grade supply.
FAQ
What is aseptic processing in simple terms?
Aseptic processing is the practice of sterilizing a product and its packaging separately, then combining them in a controlled sterile environment to prevent contamination. It is the standard method for manufacturing sterile pharmaceuticals and shelf-stable food products.
Why is aseptic processing required for biologics?
Biologics including monoclonal antibodies and cell therapies are heat-sensitive and cannot survive terminal sterilization without losing potency or structural integrity. Aseptic fill-finish is the only manufacturing method that preserves efficacy while achieving the sterility required for injectable products.
How does aseptic processing differ from terminal sterilization?
Terminal sterilization applies heat, radiation, or chemicals to the final sealed product, while aseptic processing sterilizes components separately and assembles them in a sterile environment. Aseptic methods are required when the product itself cannot tolerate the conditions needed for terminal sterilization.
What is the biggest risk in aseptic manufacturing?
The aseptic transfer step, where product or materials cross a sterile boundary, carries the highest contamination risk. Precise control of pressure differentials, physical transitions, and barrier integrity at these points is critical to maintaining sterility throughout the process.
How long do aseptically processed products last?
Aseptically filled food and beverage products typically achieve 12-18 months of ambient shelf life, confirmed through accelerated and real-time quality testing. Pharmaceutical products vary based on formulation, but the method enables room-temperature storage for many products that would otherwise require refrigeration.


